MeCP2 mutaties
Tip
Om de onderstaande tekst goed te begrijpen adviseer ik u eerst de pagina's van genoom naar basen en MeCP2 te lezen.
Genotype - Fenotype
Het is bekend dat het Rett-syndroom veroorzaakt wordt door een mutatie van het MeCP2 gen. De mutatie is het gevolg van de genetische aanleg van een individu. Hier spreekt men van het genotype. Het totaal van alle waarneembare eigenschappen, de kenmerken, noemt men het fenotype.
Om dit naar Rett te vertalen betekent het dus dat voor de diagnose Rett-syndroom alleen gekeken wordt naar het fenotype (diagnostische criteria) en dat bij een klinisch genetisch onderzoek alleen gekeken wordt naar het al dan niet aanwezig zijn van een MeCP2 gerelateerde mutatie. Het is dus mogelijk om het Rett-syndroom te hebben zonder MeCP2 gerelateerde mutatie en ook is het mogelijk om een MeCP2 gerelateerde mutatie te hebben zonder Rett-syndroom.
Meer informatie over de diagnostische criteria en de kenmerken kunt u lezen op de pagina kenmerken. Maar die kenmerken, het fenotype, worden natuurlijk veroorzaakt door de mutatie, het genotype. Over die relatie ga ik proberen meer uitleg te geven.
Bron: wikipedia.org/Fenotype
Let op!
Wat ik hieronder schrijf wordt niet ondersteunt door (alle) artsen in Nederland. Ik wil wel benadrukken dat de patiëntgroep in Nederland te klein is om hier uitgebreid onderzoek naar te doen. Waarbij dus een rol speelt dat de arts die u spreekt weinig kinderen ziet, zeker als het niet om de hotspot-mutaties gaat, om een goed oordeel te geven. Ik heb mails gestuurd naar in het buitenland zeer gerenommeerde artsen en wetenschappers en wat die aangeven staat haaks op wat ik in Nederlandse ziekenhuizen en zelfs het expertisecentrum te horen heb gekregen. Wat ik hieronder schrijf is gebaseerd op buitenlands onderzoek, waar wel grotere patiëntgroepen beschikbaar zijn voor wetenschappelijk onderzoek. Beslis waar u zich comfortabel bij voelt. Als ik het kort samenvat is wat ik in Nederland te horen krijg is vooral ‘Stop met zoeken, accepteer het syndroom van Rett. Wat u ook vindt, het zal voor uw dochter niet anders worden’. Hier zit een kern van waarheid in, maar ik ben van mening dat het probleem begrijpen de weg naar de oplossing is. En oplossing is een breed begrip. Wat is dan de oplossing? Genezing? Ja natuurlijk, maar ook symptomen in een vroeg stadium herkennen, weten waardoor ze ontstaan en over een kansberekening beschikken voor bepaalde ernstige kenmerken draagt al bij als ik mij afvraag hoe het leven van mijn dochter er later uit gaat zien. Want ook al wil de arts hier niets over bevestigen, er is een relatie tussen fenotype (uiting) en genotype (mutatie). Er is onderzoek naar gedaan, ook recentelijk, en ik wil u die informatie niet onthouden.
3 factoren bepalen de ernst
Er zijn 3 factoren die een rol spelen bij de uiting (fenotype) van het syndroom van Rett.
1. Het type mutatie en de locatie
2. Een scheef XCI-patroon
3. De aanwezigheid van modificerende genen
Bron: link.springer.com/Treating Rett syndrome: from mouse models to human therapies
1a. Het type mutatie
Er zijn verschillende soorten mutaties, waarbij de meestvoorkomende missense mutaties, nonsense mutaties en frameshift mutaties (waaronder inserties en deleties) zijn. Ik ga, aan de hand van voorbeelden, u eenvoudig uit proberen te leggen wat deze mutaties precies inhouden.
Bron: ghr.nlm.nih.gov/possiblemutations
Missense mutatie
Een missense mutatie wordt, net als de nonsense mutatie, vaak veroorzaakt door een puntmutatie in het DNA. Bij een puntmutatie wordt één enkele nucleotide (base) vervangen door een andere nucleotide. De wijziging van deze nucleotide, ook wel substitutie genoemd, leidt bij een missense mutatie tot het inbouwen van een ander aminozuur in het eiwit. Door het inbouwen van een ander aminozuur verliest het eiwit soms niet helemaal zijn functionaliteit.
Voorbeeld
Als we even het voorbeeld van het boek erbij pakken helemaal onderaan op de pagina van genoom naar basen, dan staan er in dat boek (het genoom) allemaal 3-letter woorden, waaronder de zin ‘zij had een kat’. Bij een puntmutatie kan de ‘k’ vervangen worden door een ‘r’, dan staat er ‘zij had een rat’. Dat is een normale kloppende zin, alleen met een iets andere betekenis. Maar de ‘k’ kan ook vervangen worden door een ‘w’, dan staat er ‘zij had een wat’. En dat is weer een zin die niet klopt.
Bron: biologielessen.nl/missence-mutatie
Bron: www.spierziekten.nl/mutaties-en-ziekten

Afbeelding: US National Library of Medicine
Nonsense mutatie
Een nonsense mutatie, wat letterlijk onzin mutatie betekent, wordt net als een missense mutatie veroorzaakt door een puntmutatie in het DNA. Echter, in plaats van een vervanging van het ene aminozuur door het andere, is bij een missense mutatie sprake van een stopcodon. De hierdoor veranderde DNA sequentie geeft de cel vroegtijdig een signaal om te stoppen met het bouwen van een eiwit. Dit type mutatie resulteert in een tekort eiwit dat mogelijk niet of slecht functioneert.
Voorbeeld
Als we even het voorbeeld van het boek erbij pakken helemaal onderaan op de pagina van genoom naar basen, dan staan er in dat boek (het genoom) allemaal 3-letter woorden, waardoor de zin ‘zij had een kat.’. Bij een nonsense mutatie staat er ‘zij had een.’. De punt, die normaal achter ‘kat’ staat, staat nu achter ‘een’, waardoor de zin vroegtijdig afgebroken wordt.
Bron: ghr.nlm.nih.gov/possiblemutations
Bron: spierziekten.nl/mutaties-en-ziekten

Afbeelding: US National Library of Medicine
{kind=link}
Frameshift mutatie
Een frameshift mutatie treedt op wanneer de toevoeging of het verlies van DNA-basen het leesraam van een gen verandert. Een leesraam bestaat uit telkens 3 basen (codons) die elk coderen voor één aminozuur. Een frameshift mutatie verschuift de groepering van deze basen en verandert de code voor aminozuren na de mutatie. Een frameshift mutatie ontstaat wanneer het totaal aantal ingevoegde nucleotiden niet deelbaar is door 3 (het aantal basen per codon). De frameshift mutatie zorgt er in de regel voor dat de actieve translatie van gen voortijdig een stopcodon tegenkomt, wat resulteert dus niet resulteert in een correct eiwitproduct. Het eiwit wat door dit gen geproduceerd wordt zal vanaf de mutatie geheel veranderd zijn en meestal geen functie meer kunnen vervullen. Men spreekt dat van een truncerende mutatie (afgeknot of ingekort eiwit). Inserties en deleties, die samen ook wel InDel's genoemd worden, zijn mutaties die een frameshift tot gevolg kunnen hebben.
Insertie (die leidt tot een frameshift mutatie)
Bij een insertie verandert het aantal DNA-basen in een gen door een stukje DNA toe te voegen. Als gevolg hiervan werkt het eiwit dat door het gen wordt gemaakt mogelijk niet goed.
Voorbeeld
Als we even het voorbeeld van het boek erbij pakken helemaal onderaan op de pagina van genoom naar basen, dan staan er in dat boek (het genoom) allemaal 3-letter woorden, waaronder de zin ‘zij had een kat’. Bij een insertie wordt er bijvoorbeeld 1 letter toegevoegd en wordt de zin onleesbaar, bijvoorbeeld ‘zij aha dee nka t’, wanneer een ‘a’ voor 'had' geplaatst wordt.
Bron: ghr.nlm.nih.gov/possiblemutations
Bron: wikipedia.org/Frameshiftmutatie
Bron: wikipedia.org/Insertion (genetics)
Bron: spierziekten.nl/erfelijkheid/mutaties-en-ziekten

Deletie (die leidt tot een frameshift mutatie)
Bij een deletie verandert het aantal DNA-basen in een gen door een stukje DNA te verwijderen. Als gevolg hiervan werkt het eiwit dat door het gen wordt gemaakt mogelijk niet goed.
Voorbeeld
Als we even het voorbeeld van het boek erbij pakken helemaal onderaan op de pagina van genoom naar basen, dan staan er in dat boek (het genoom) allemaal 3-letter woorden, waaronder de zin ‘zij had een kat’. Bij een deletie wordt er bijvoorbeeld 1 letter verwijderd en wordt de zin onleesbaar, bijvoorbeeld ‘zij ade enk at’, wanneer de ‘h’ bij ‘had’ verwijderd wordt.
Bron: ghr.nlm.nih.gov/possiblemutations
Bron: spierziekten.nl/mutaties-en-ziekten

Afbeelding: US National Library of Medicine
1b. De locatie van de mutatie
Een mutatie kan op verschillende locaties aanwezig. Allereerst is het domein waar de mutatie zich bevindt belangrijk. Wat deze domeinen precies doen kunt u lezen op de pagina MeCP2. Er wordt vanuit gegaan dat, nog even los van de exacte mutatie, het fenotype ernstiger is naarmate de mutatie zich meer bevindt op de eerste helft van het MeCP2 gen. Dit is globaal genomen, los even van de exacte mutatie want die is natuurlijk leidend voor de ernst van symptomen.

Afbeelding: The Journal of Clinical Investigation
8 hotspot mutaties
Bij grotere onderzoeken onder Rett-patiënten is gebleken dat veel patiënten dezelfde mutaties hebben. Deze veelvoorkomende mutaties noemt men de hotspots. Bij circa 68% van de Rett-patiënten is een mutatie op één van deze hotspots de veroorzaker van het Rett-syndroom. Dit is gebleken uit verzamelde data van de internationale Rett syndroom fenotype database InterRett, die gecombineerd werd met de grote Australian Rett Syndrome Database (ARSD).
Bron: ncbi.nlm.nih.gov/pmc/The phenotype associated with a large deletion on MECP2
De hotspot mutaties zijn:
R106W / Arg106Trp = missense mutatie
Uitgeschreven staat er dat er op positie 106 een arginine (aminozuur) zou moeten zijn om een gezond eiwit aan te maken maar bij de patiënt een tryptofaan (aminozuur) aanwezig is. Deze mutatie komt voor bij ca. 4% van de Rett patiënten. Uit onderzoeken onder patiënten is niet gebleken dat het fenotype relatief milder of ernstiger is dan andere mutaties.
R133C / Arg133Cys = missense mutatie
Uitgeschreven staat er dat er op positie 133 een arginine (aminozuur) zou moeten zijn om een gezond eiwit aan te maken maar bij de patiënt een cysteine (aminozuur) aanwezig is. Deze mutatie komt voor bij ca. 7% van de Rett patiënten. Uit onderzoeken onder patiënten is gebleken dat de R133C mutatie relatief een milder fenotype kent. De reden hiervoor is dat wordt aangenomen dat ondanks de R133C mutatie, het MBD domein nog gedeeltelijk zijn functie houdt.
T158M / Thr158Met = missense mutatie
Uitgeschreven staat er dat er op positie 158 een threonine (aminozuur) zou moeten zijn om een gezond eiwit aan te maken maar bij de patiënt een methionine (aminozuur) aanwezig is. Deze mutatie komt voor bij ca. 13% van de Rett patiënten en is de meest voorkomende mutatie. Uit onderzoeken onder patiënten is niet gebleken dat het fenotype relatief milder of ernstiger is dan andere mutaties.
R168X / Arg168X = nonsense mutatie
Uitgeschreven staat er dat er op positie 168 een arginine (aminozuur) zou moeten zijn om een gezond eiwit aan te maken maar bij de patiënt de eiwitsynthese stopt door een vroegtijdig (prematuur) stopcodon. Hierdoor ontstaat een tekort (afgeknot) eiwit. Deze mutatie komt voor bij ca. 11% van de Rett patiënten. Uit onderzoeken onder patiënten is gebleken dat de R168X mutatie, gelegen in het tussenliggende domein ID, nog voor het TRD domein, een relatief ernstiger fenotype kent.
R255X / Arg255X = nonsense mutatie
Uitgeschreven staat er dat er op positie 255 een arginine (aminozuur) zou moeten zijn om een gezond eiwit aan te maken maar bij de patiënt de eiwitsynthese stopt door een vroegtijdig (prematuur) stopcodon. Hierdoor ontstaat een tekort (afgeknot) eiwit. Deze mutatie komt voor bij ca. 11% van de Rett patiënten. Uit onderzoeken onder patiënten is niet gebleken dat het fenotype relatief milder of ernstiger is dan andere mutaties.
R270X / Arg270X = nonsense mutatie
Uitgeschreven staat er dat er op positie 270 een arginine (aminozuur) zou moeten zijn om een gezond eiwit aan te maken maar bij de patiënt de eiwitsynthese stopt door een vroegtijdig (prematuur) stopcodon. Hierdoor ontstaat een tekort (afgeknot) eiwit. Deze mutatie komt voor bij ca. 8% van de Rett patiënten. Uit onderzoeken onder patiënten is gebleken dat de R270X mutatie, gelegen in de belangrijke TRD-NLS regio van het TRD domein, een relatief ernstiger fenotype kent.
R294X / Arg294X = nonsense mutatie
Uitgeschreven staat er dat er op positie 294 een arginine (aminozuur) zou moeten zijn om een gezond eiwit aan te maken maar bij de patiënt de eiwitsynthese stopt door een vroegtijdig (prematuur) stopcodon. Hierdoor ontstaat een tekort (afgeknot) eiwit. Deze mutatie komt voor bij ca. 7% van de Rett patiënten. Uit onderzoeken onder patiënten is gebleken dat de R294X mutatie relatief een milder fenotype kent. De reden hiervoor kan de locatie zijn in het laatste stuk van het TRD domein, net na de belangrijk TRD-NLS regio, waardoor het resulterende eiwit mogelijk nog deels werkzaam is.
R306C / Arg306Cys = missense mutatie
Uitgeschreven staat er dat er op positie 306 een arginine (aminozuur) zou moeten zijn om een gezond eiwit aan te maken maar bij de patiënt een cysteine (aminozuur) aanwezig is. Deze mutatie komt voor bij ca. 7% van de Rett patiënten. Uit onderzoeken onder patiënten is gebleken dat de R306C mutatie relatief een milder fenotype kent. De reden hiervoor kan de locatie zijn in het laatste stuk van het TRD domein, na de belangrijk TRD-NLS regio, waardoor het resulterende eiwit mogelijk nog deels werkzaam is.
Bron: researchgate.net/Location-and-frequency-of-MeCP2-hotspot-mutations-in-RTT-patients
Bron: ncbi.nlm.nih.gov/Rett Syndrome-causing Mutations in Human MeCP2 Result in Diverse Structural Changes That Impact Folding and DNA Interactions
Bron: jmg.bmj.com/Refining the phenotype of common mutations in Rett syndrome
Bron: informatics.jax.org/allele/Mecp2

Afbeelding: Researchgate.net
Bron: researchgate.net/Location-and-frequency-of-MeCP2-hotspot-mutations-in-RTT-patients
Vroege truncaties, grote deleties en C-terminale deleties
Door de data van de internationale Rett syndroom fenotype database te combineren met de grote Australian Rett Syndrome Database (ARSD) bleek dat, naast de 68% patiënten met mutaties op de hotspots, er ook vaak sprake was van vroege truncaties (5%), grote deleties (5%) en C-terminale deleties (10%). Samen met de hotspots dekt dit 88% van alle patiënten met Rett Syndrome uit de gecombineerde database.
Vroege truncaties
Onder vroege truncaties worden de nonsense mutaties verstaan voor aminozuur 310, met uitzondering van de hotspot nonsense mutaties R168X, R255X, R270X en R294X. Wanneer er een te kort (afgeknot of ingekort) eiwit wordt aangemaakt, meestal veroorzaakt door een vroegtijdig stopcodon, dan spreekt men van een truncerende mutatie. Het eiwit is dan vanaf de mutatie geheel veranderd en kan meestal geen functie meer vervullen. Deze soort mutatie komt voor bij ca. 5% van de Rett patiënten. Uit onderzoeken onder patiënten is gebleken dat vroege truncerende mutaties, gelegen in de belangrijke domeinen regio van het MeCP2, een relatief ernstiger fenotype kennen.
Grote deleties
Onder grote deleties worden grote missende delen verstaan met uitzondering van deleties in het C-terminale domein. Uit het onderzoek is gebleken dat de meeste grote deleties gevonden worden in exons 3 en 4 van het MeCP2 gen. Bij grote deleties is er vrijwel altijd sprake van een frameshift met als gevolg dat er bij het coderen een vroegtijdig stopcodon gevonden wordt. Hierdoor wordt er een te kort (afgeknot of ingekort) eiwit aangemaakt. Het eiwit is dan vanaf de mutatie geheel veranderd en kan meestal geen functie meer vervullen. Deze soort mutatie komt voor bij ca. 5% van de Rett patiënten. Uit onderzoeken onder patiënten is gebleken dat grote deleties, meestal gelegen in de belangrijke exonen 3 en 4 van het MeCP2, relatief gezien het meest ernstige fenotype kennen van alle mutaties.
C-terminale deleties
Hoewel grote deleties relatief een ernstiger fenotype kennen is dat anders bij C-terminale domein deleties, gelegen op laatste stuk van exon 4 (aminozuren vanaf 310) van het MeCP2 gen. Deze mutatie komt voor bij ca. 10% van de Rett patiënten. Uit onderzoeken onder patiënten is gebleken dat C-terminale deleties, net als de hotspot mutatie R133C, relatief een milder fenotype kennen. Door deze mildheid is besloten deze groep deleties apart te definiëren. Er is meer onderzoek nodig onder grotere groepen Rett-patiënten met C-terminale domein mutaties om meer te weten te komen over deze recent geïdentificeerde mutatiegroep.
Bron: ncbi.nlm.nih.gov/The phenotype associated with a large deletion on MECP2
Andere mutaties
Naast de 8 hotspot mutaties zijn er natuurlijk nog vele andere mutaties. Hieronder een kort overzicht van andere mutaties die ik op wat voor manier dan ook tegengekomen ben. Het overzicht is niet compleet. U mag daarom altijd een mutatie doorgeven en ik zal kijken wat ik erover kan vinden. Vanzelfsprekend doe ik aan bronvermelding zodat u altijd kunt zien waar de geplaatste informatie vandaan komt.
L336P / Leu336Pro = C-terminale deletie (Dochter Nikki)
Bij onze dochter Nikki is sprake van een C-terminale deletie, namelijk c.1007_1194del(p.Leu336Profs*6). Dit betekent dat er op positie 336 een Leucine (Leu of L) aanwezig zou moeten zijn om een gezond eiwit aan te maken. Echter is op die locatie een Proline (Pro of P) aanwezig, wat resulteert in de mutatie Leu336Pro (of L336P). Dit is het gevolg van de deletie (c.1007_1194del188), waardoor nucleotide 1006 een codon vormt met nucleotiden 1195 en 1196. Er missen dus 188 nucleotiden, oftewel op-één-nucleotide-na 63 aminozuren. Hierdoor wordt gecodeerd voor Proline in plaats van Leucine. Door deze deletie ontstaat een frameshift (hiervoor staan de letters fs in de mutatie) en kloppen de daaropvolgende aminozuren niet waarna het aflezen stopt door een vroegtijdig stopcodon bij positie 6 (*6). Onze dochter Nikki heeft naar het nu lijkt een milde vorm van het Rett Syndroom, omdat zij veel van de ernstige kenmerken niet heeft. Oorzaak van deze mildheid kan zijn dat de mutatie in het C-terminale domein ligt. Uit onderzoeken onder patiënten is gebleken dat C-terminale deleties relatief een milder fenotype kennen. De mutatie van Nikki is nergens beschreven en is, zo is ons door LUMC verteld, niet eerder gemeld in de wereldwijde database.
S134C / Ser134Cys = missense mutatie
Uitgeschreven staat er dat er op positie 134 een serine (aminozuur) zou moeten zijn om een gezond eiwit aan te maken maar bij de patiënt een cysteine (aminozuur) aanwezig is. Het inbouwen van een ander aminozuur is het gevolg van een missense mutatie, ook wel puntmutatie genoemd (c.401C>G). Bij deze puntmutatie wordt één enkele nucleotide (C) vervangen door een andere nucleotide (G). Uit onderzoeken onder patiënten is gebleken dat de S134C mutatie relatief een milder fenotype kent, net als de naastgelegen R133C mutatie. De reden hiervoor is dat wordt aangenomen dat ondanks de S134C mutatie, het MBD domein nog gedeeltelijk zijn functie houdt.
Bron: ncbi.nlm.nih.gov/clinvar/NM_004992.3(MECP2):c.401C>G (p.Ser134Cys)
Bron: jmg.bmj.com/content/Heterogeneity in residual function of MeCP2 carrying missense mutations in the methyl CpG binding domain
P389X / Pro389X = C-terminale deletie
Pro389X is een C-terminale deletie, namelijk c.1164_1207del(p.Pro389*). Dit betekent dat er op positie 389 een Proline (Pro of P) aanwezig zou moeten zijn om een gezond eiwit aan te maken. Echter is op die locatie een vroegtijdig stopcodon, wat resulteert in de mutatie Pro389X (of P389X). Dit is het gevolg van de deletie (c.1164_1207del44), waardoor nucleotiden 1162 en 1163 een codon vormen met nucleotide 1208. Er missen dus 44 nucleotiden, oftewel op-één-nucleotide-na 15 aminozuren. Hierdoor wordt gecodeerd voor een stopcodon in plaats van Proline. Uit onderzoeken onder patiënten is gebleken dat de C-terminale domein mutaties relatief een milder fenotype kennen. De reden hiervoor is dat wordt aangenomen dat het MBD domein nog gedeeltelijk zijn functie houdt.
Bron: dspace.library.uu.nl/Gastroschisis; perinatale en postnatale aspecten
Bron: mecp2.chw.edu.au/mecp2/MECP2 Variant List

Dit is de tabel met de data van InterRett en de Australian Rett Syndrome Database (ARSD) die gebruikt zijn voor het onderzoek.
InterRett is opgericht in 2003 en bevat onder meer data van Rettnett en ingestuurde vragenlijsten door families en clinici. Dit zijn gegevens uit Australië, maar ook uit o.a. Spanje, Frankrijk, Israël en Canada.
De ARSD database is uniek omdat hier vrijwel alle Australische Rett-patiënten geboren sinds 1976 in staan. Sinds 2000 wordt iedere 2-3 jaar alle gegevens van deze patiënten gecollecteerd.
Bron en afbeelding: ncbi.nlm.nih.gov/The phenotype associated with a large deletion on MECP2

Dit is het overzicht van ernstscores (severity score) van alle hotspot mutaties, aangevuld met de C-terminale deleties, vroege truncaties, grote deleties en overig.
Op de groep Rett patiënten zijn de criteria van Pineda (Pineda score, 776 patiënten) en criteria van Percy (Percy score, 974 patiënten) toegepast. Dr. Percy is de oprichter van het Rare Disease Consortium Research Network for RTT, opgericht in 2004 in de VS. Dr Pineda uit Spanje (Monrós et al., 2001) heeft een meetmethode ontwikkeld om een reeks symptomen te meten a.d.h.v. klinische ernstscores.
Bron en afbeelding: rettuk.org/Clinical and biological progress over 50 years in Rett syndrome

Op bovenstaande afbeelding is te zien dat patiënten met de mildere varianten, waaronder R133C (Arg133Cys), R294X (Arg294X), C-terminale deleties en R306C (Arg306Cys) relatief op latere leeftijd gediagnosticeerd worden. Dit is vaak een gevolg van het ontbreken van essentiële kenmerken, waardoor niet in eerste instantie aan Rett Syndroom gedacht wordt.
Deze data is afkomstig van InterRett en de Australian Rett Syndrome Database (ARSD). Het is gebaseerd op een onderzoek onder 1040 patiënten met Rett Syndroom.
Bron en afbeelding: rettuk.org/Clinical and biological progress over 50 years in Rett syndrome



Afbeeldingen A, B en C: Functionele vaardigheden
Deze grafieken tonen de relatie tussen de MeCP2 mutatie en het functioneel vermogen bij patiënten met het Rett Syndroom. Gegevens zijn afkomstig van InterRett. Grafiek ambulatievermogen (A) is gebaseerd op data van 1112 patiënten, grafiek handgebruik (B) is gebaseerd op data van 1097 patiënten en grafiek taalvaardigheid (C) is gebaseerd op data van 1046 patiënten.
Bron en afbeelding: rettuk.org/Clinical and biological progress over 50 years in Rett syndrome

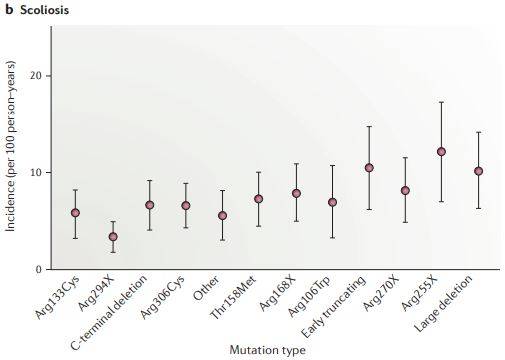
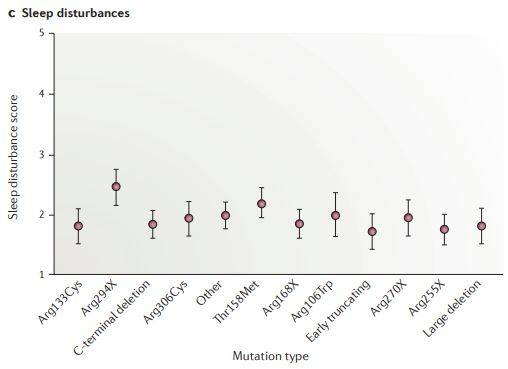
Afbeeldingen A, B en C: epilepsie, scoliose en slaapstoornissen
Deze grafieken tonen de relatie tussen de MeCP2 mutatie en de veel bij Rett patiënten voorkomende kenmerken epilepsie, scoliose en slaapstoornissen.
Grafiek epilepsie (A) is gebaseerd op data van 560 patiënten, grafiek scoliose (B) is gebaseerd op data van 392 personen en grafiek slaapstoornissen (C) is gebaseerd op data van 325 patiënten.
Bron en afbeelding: rettuk.org/Clinical and biological progress over 50 years in Rett syndrome
2. Een scheef XCI-patroon
Wanneer er sprake is van een scheef XCI-patroon kan dit zorgen voor een milder fenotype of een ernstiger fenotype. Hieronder leg ik uit wat een scheef XCI-patroon precies inhoudt.
X-chromosomen
Mannen hebben een X en een Y als geslachtschromosomen terwijl vrouwen twee X-chromosomen hebben. In elke lichaamscel is één chromosoom altijd uitgeschakeld, want met twee inschakelde X-chromosomen is leven niet mogelijk.
Bron: ru.nl/uitzetten-chromosoom-vrouwen-beeld-gebracht
X-inactivatie
X-inactivatie, ook wel XCI (X-chromosome inactivation) genoemd, is een proces dat ervoor zorgt dat de genen van één van de twee X-chromosomen bij vrouwen tijdens de embryonale ontwikkeling uitgeschakeld worden, zodat ze net als mannen maar één actief X-chromosoom hebben. Welke van de twee uitgeschakeld wordt volledig wikkekeurig beslist (random inactivation). Gewoonlijk is het voor 50% het ene X-chromosoom, en voor 50% het andere.
Bron: ru.nl/uitzetten-chromosoom-vrouwen-beeld-gebracht
Scheef XCI-patroon
Een scheef XCI-patroon ontstaat wanneer één X-chromosoom vaker uitgeschakeld is dan de ander. Een bepaalde mate van scheefheid is normaal, 70%-30% is relatief gangbaar bij ca. 35% van de vrouwen. Bij een verhouding 75%-25% is er sprake van een scheef XCI patroon en bij een verhouding 90%-10% spreekt men van een extreem scheef XCI patroon. Een scheef XCI patroon is aanwezig bij ca. 7% van de vrouwen.
Bron: wikipedia.org/Skewed_X-inactivation

Rett Syndroom
Bij Rett-patiënten is vrijwel altijd een mutatie op het X-chromosoom de oorzaak. Aangezien mannen met een mutatie op het X-chromosoom, uitzonderingen daargelaten, niet levensvatbaar zijn, gaat men ervan uit dat er naast het X-chromosoom met de mutatie nog een gezond X-chromosoom aanwezig is. Er is theorie dat wanneer het actieve X-chromosoom een gen met een mutatie bevat, de kans bestaat dat deze cellen niet overleven gedurende de ontwikkeling en de andere cellen van het uitgeschakelde X-chromosoom het dan over nemen. Een andere theorie is het X-chromosoom met de MeCP2 mutatie vaker paternaal (van de vader) verkregen is (in ca. 80% van de gevallen), en dat het lichaam van nature een lichte voorkeur heeft de van de vader verkregen X vaker uit te schakelen.
Wanneer het ene X-chromosoom vaker uitgeschakeld is dan het andere ontstaat er een scheef XCI patroon, ook wel skewed, skewing of non-random patroon genoemd, meestal in het voordeel van het X-chromosoom met het normale gen (wild-type). Er wordt gedacht dat iets dergelijks ook plaatsvindt bij MeCP2 mutaties. In ieder geval is wel duidelijk dat een scheef XCI-patroon, waarbij de het X-chromosoom met het gemuteerde MeCP2 gen vaker uitgeschakeld is, bescherming biedt tegen de ernst van de MeCP2 mutatie. Uit onderzoek is gebleken dat de meeste geteste Rett-patiënten (ca. 90%) een normaal XCI-patroon (random) laten zien. Er zijn echter voorbeelden van patiënten die een milder fenotype lieten zien dan wat gezien de locatie van de mutatie verwacht zou worden, en waar na onderzoek een scheef XCI-patroon is gevonden.
Bron: wikipedia.org/Skewed_X-inactivation
Bron: nemokennislink.nl/uitschakelen-x-chromosoom-in-beeld-gebracht
Bron: www.rettsyndrome.be/Dossier MeCP2
Bron: www.ncbi.nlm.nih.gov/Correlation between clinical severity in patients with Rett syndrome and the direction and degree of skewing of X‐chromosome inactivation
Bron: ncbi.nlm.nih.gov/Parental origin of de novo MECP2 mutations in Rett syndrome
3. De aanwezigheid van modificerende genen
De eerste keer dat hoorde van modificerende genen was toen Adrian Bird, een bekende klinisch geneticus uit Engeland, mijn e-mail met vragen beantwoordde. Hij stuurde onderstaande tekst in zijn mail aan mij.
“Another potential reason for mildness is the presence of “modifier genes” – i.e. variants of genes other than MECP2. Each human has a somewhat different genome, with its own specific collection of polymorphisms. There is plenty of evidence that the effects of mutations (not only Rett syndrome) heavily depend on this "genetic background". The same mutation can have very different effects depending on what other gene variants that individual is carrying. At present we do not know for Rett whether a few other genes contribute to this or if is the aggregate effect of tens or hundreds of slightly different genes. There is some work in mice which pinpoints certain genes as responsible, but this knowledge does not yet suggest clinical interventions.”
De vertaling is:
“Een andere mogelijke reden voor mildheid (van Rett Syndroom, red.) is de aanwezigheid van “modificerende genen”, d.w.z. varianten van andere genen dan MeCP2. Ieder mens heeft een ander genoom, met zijn eigen specifieke verzameling polymorfie (variaties in DNA, red.). Er zijn voldoende aanwijzingen dat de effecten van mutaties (niet alleen voor het Rett-syndroom) sterk afhankelijk zijn van deze “genetische achtergrond”. Dezelfde mutatie kan zeer verschillende effecten hebben, afhankelijk van welke andere genvarianten die persoon draagt. Op dit moment weten we voor Rett niet of een paar andere genen hieraan bijdragen of dat het het geaggregeerde (gecombineerde, totale, red.) effect is van tientallen of honderden enigszins verschillende genen. Er is wat werk bij muizen dat bepaalde genen als verantwoordelijk identificeert, maar deze kennis suggereert nog geen klinische interventies.”
Bron: e-mail van Adrian Bird als antwoord op vragen over mijn dochter Nikki.
Adrian Bird
Sir Adrian Peter Bird (geboren 3 juli 1947) is een Britse geneticus en Buchanan hoogleraar genetica aan de Universiteit van Edinburgh. Hij heeft een grote rol gespeeld bij het opzetten van het Welcome Trust Center for Cell Biology, waarvan hij directeur was van 1999 tot 2011. Adrian Bird was van 2000 tot 2010 gouverneur van Welcome Trust, 's werelds grootste liefdadigheidsinstelling voor medisch onderzoek en verbonden aan Cancer Research UK.
Adrian Bird is voormalig voorzitter van de Scientific Advisory Board van Rett Syndrome Research Trust (RSRT). Hij is 's werelds toonaangevende expert in het MeCP2 gen, veroorzaker van het Rett-syndroom. Bird's groep, ook wel het Bird Lab genoemd, ontdekte dat het MeCP2 eiwit specifiek bindt aan gemethyleerde CpG-eilanden. In 2007 publiceerde het Bird-laboratorium in het tijdschrift Science een stuk waarin een experiment werd beschreven waarbij het Rett-syndroom bij laboratoriummuizen met succes kon worden teruggedraaid. Dit werd bereikt door het terugplaatsen van een functioneel MeCP2-gen en bleek succesvol, zelfs wanneer de toestand zich in een vergevorderd stadium bevond. Dit wijst op de mogelijkheid van gentherapie om Rett-syndroom in de toekomst te genezen.
Adrian Bird is in 2005 benoemd tot 'Commander of the Order of the British Empire'. In 2013 is Adrian Bird getipt als potentiële winnaar van de Nobelprijs voor fysiologie en geneeskunde in 2013, maar heeft deze prijs niet gekregen. Verder heeft Adrian Bird de Buchanan Medal of the Royal Society ontvangen in 2018 voor zijn medische ontdekkingen.
Bron: wikipedia.org/Adrian Bird
Bron: reverserett.org/sir-adrian-p-bird
Muismodellen
Muismodellen zijn een essentieel hulpmiddel voor het bestuderen van het Rett-syndroom. De gevolgen van een MeCP2-tekort bij muizen sluiten nauw aan bij de klinische kenmerken van de menselijke aandoening. Zo hebben muizen ook motorische defecten en ademhalingsstoornissen. Bij een studie onder dieren waarbij 3 MeCP2-mutaties verschillende klinische ernst tot expressie brachten toonden de muizen het meest passende ernstspectrum. Deze bevindingen bevestigen de overeenkomst van de MeCP2-aminozuursequentie tussen mens en muis (95% identiek) en de parallelle dynamiek van MeCP2 ontwikkeling van de hersenen. Ook al verschillen mensen en muizen in hersenstructuur en ontwikkelingstiming, er zijn opvallende overeenkomsten dat de moleculaire gevolgen van een MeCP2 mutatie vergelijkbaar zijn tussen de twee soorten.
Bron: ncbi.nlm.nih.gov/MeCP2 mutations: progress towards understanding and treating Rett syndrome
Wat zijn modificerende genen?
Modificerende genen zijn genen naast het hoofdgen¹. De werking van een gen dat meer dan één kenmerk beïnvloedt heet pleiotropie. Meestal wordt een kenmerk dat een kwantitatieve variabiliteit vertoont bepaald door de samenwerking van meerdere genparen (polygenen), die ieder verantwoordelijk zijn voor een klein effect². Binnen ieder genpaar kan de werking intermediair of dominant zijn. Een genetische eigenschap is intermediair als beide allelen (kopieën) van een gen, zowel het allel op het chromosoom afkomstig van de moeder als het allel afkomstig van het chromosoom van de vader, tot uiting (expressie) komen. Hierdoor is het fenotypische kenmerk een mengvorm van beide allelen³. Een genetische eigenschap is dominant aanwezig als één van de twee allelen (kopieën) van een gen, of die op het van de moeder afkomstige chromosoom of die op het van de vader afkomstige chromosoom, tot uiting (expressie) komt. Het niet tot expressie komende allel heet recessief⁴. Wanneer verschillende genen elkaar versterken (additief werken) spreekt men van cumulatieve polygenie of homomerie. Cumulatief samenwerkende genen hebben vaak een werkingsgraad van zeer verschillende sterkte, zodat men naast een hoofdgen, dat de ontwikkeling van een kenmerk voornamelijk bepaalt, nog als modificatoren optredende bijgenen of modificerende genen kan onderscheiden. Deze staan meestal samen als het onverdeelde restgenotype tegenover één paar hoofdgenen².
¹Bron: woordjesleren.nl/modificerende genen
²Bron: jkwiersema.nl/begrippen uit de erfelijkheidsleer
³Bron: wikipedia.org/Intermediair_(genetica)
⁴Bron: wikipedia.org/Dominant_(genetica)
Modificerende mutaties
Wanneer Rett-patiënten met exact dezelfde mutatie een verschillende fenotype laten zien, kunnen deze verschillen te wijten zijn aan verschillen in X-chromosoominactivatie (een scheef XCI-patroon). Bij Rett-patiënten met dezelfde mutatie waarbij het XCI-patroon niet scheef is, maar er wel variatie in fenotype is, kan dit het gevolg zijn van de aanwezigheid van modificatiemutaties. Modificatoren zijn genen waarvan de functie fenotypische uitkomsten heeft op het effect van een ander gen. Mutaties in modificerende genen kunnen klinische symptomen bij patiënten onderdrukken (suppressor mutations) of versterken (enhancer mutations) ¹.
¹Bron: link.springer.com/Treating Rett syndrome: from mouse models to human therapies

Monica Justice
Het Rett Syndrome Research Trust (RSRT) van Monica Coenraads heeft $ 2.300.000 (ca. € 2.073.000) toegekend aan Monica Justice, PhD, Hospital for Sick Children (Toronto). Monica Justice is hoofd en senior wetenschapper van het Genetics & Genome Biology program van het Hospital for Sick Children. Zij doet onderzoek naar variaties in genetische samenstelling van kinderen die invloed hebben op de ernst van de Rett-mutatie. Zij heeft een manier ontwikkeld om deze modificerende genen te identificeren, en legt daarbij focus op onderdrukkers van symptomen, in de hoop dat het leidt tot therapeutische mogelijkheden. De eerste onderdrukker (suppressor) die ze identificeerde, squaleenepoxidase, heeft geleid tot een klinische proef met lovastatine. Monica Justice heeft tot op heden enkele tientallen modificatoren geïdentificeerd¹.
¹Bron: reverserett.org/Identification of Gene Modifiers that Ameliorate Rett Symptoms